Variant Call Format or VCF is an efficient and nonredundant way of storing gene sequence variations in text files. Any i2b2 client site that wants to use this data inside i2b2 to query for genotyped subjects will require an ETL process to convert raw VCF file data into i2b2 queryable format. The ETL process shall parse out variant annotations from the raw data in VCF files, transform, and store in the observation_blob field of the observation_fact table in i2b2 data-mart.
In the genomics package, we have provided a sample ETL .net application that can parse zipped vcf (using using gunzip library only) files with genetic variants discovered by the Illumina Multi-Ethnic Genotyping Array and load them on i2b2 SQL Server database (only). We have provided a sample zipped vcf file too. The .net application, sample vcf file and required SQL Server scripts can be found inside “ETL Process” folder inside the package. To test this application with the sample vcf file, the following steps should be followed:
- Add GENOMICS_UPLOAD_LOG table in the CRC database (the create script can be found in the package). This table is mainly used by the application to keep track of all the vcf files and patients loading status.
- Add a nullable column of integer type named SUBJECT_ID in PATIENT_DIMENSION table (Alter script can be found in the package). This column is supposed to contain patients’ local site id for a client site. For the application to work one test row in the PATIENT_DIMENSION table should be altered to have subject_id = 1000025.
- Update “MEGA_V1_B1_ETL.exe.config” file inside “i2b2_Genomics_ETL> i2b2_Genomics_ETL>bin> Debug” folder to point to local environment.
- Execute the app by double clicking on “MEGA_V1_B1_ETL.exe” inside “i2b2_Genomics_ETL> i2b2_Genomics_ETL>bin> Debug” folder that brings the following screen:
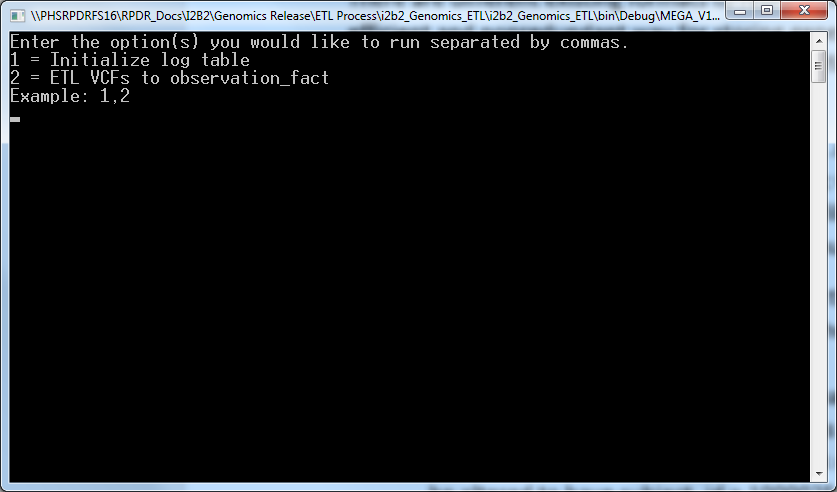
- Enter 1,2 and press enter which bring s the following screen:
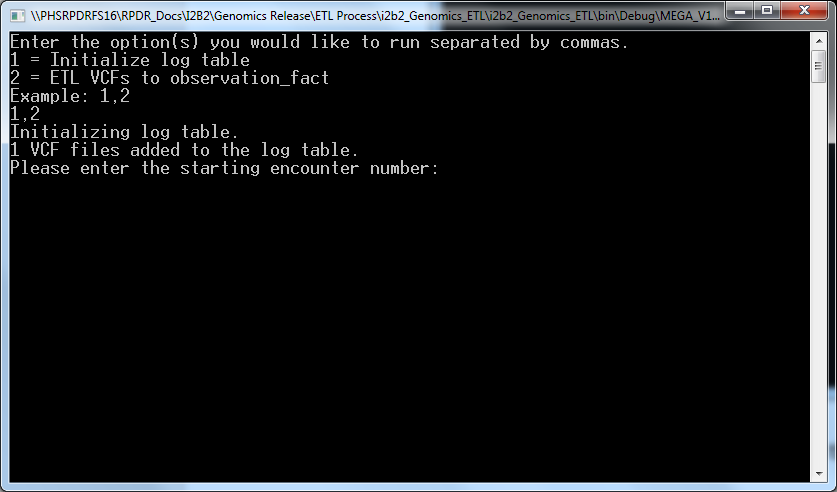
- Enter an encounter number that is more than the maximum encounter number in your CRC database and confirm that by “Y”
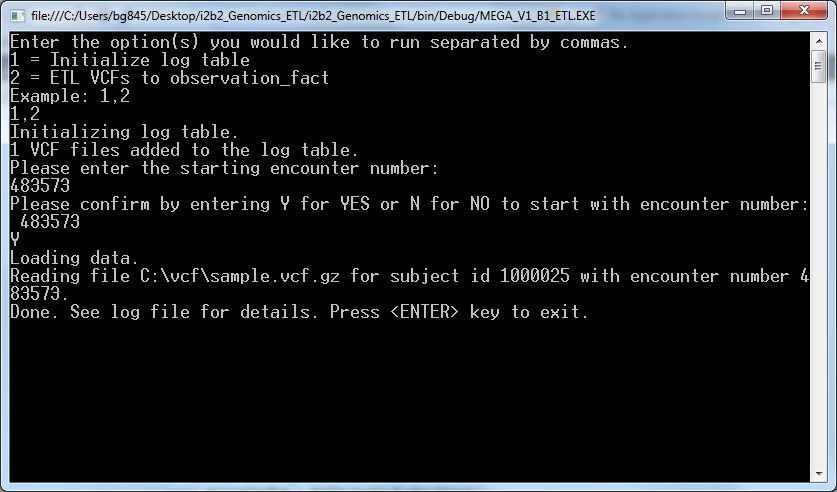
Once the process ends successfully the newly loaded data can be found in the observation fact table.
Genotype data in the observation_fact table | |||
CONCEPT_CD | INSTANCE_NUM | VALTYPE_CD | OBSERVATION_BLOB |
SO:0001483 | 1 | B | rs377573539,T_to_C,MIR6723,homozygous_ref_ref,upstream |
SO:0001483 | 2 | B | rs6429759,C_to_T,AGMAT,homozygous_alt_alt,intron |
SO:0001483 | 3 | B | rs2298948,T_to_C,GCFC2,heterozygous_ref_alt,intron |
SO:0001483 | 4 | B | rs12640778,C_to_T,LINC01060,heterozygous_ref_alt,intron |
SO:0001483 | 5 | B | rs1060583,G_to_A,NECAB1,heterozygous_ref_alt,3'UTR |
SO:0001483 | 6 | B | rs533612,A_to_G,SIK2,homozygous_alt_alt,3'UTR |
SO:0001483 | 7 | B | rs4983407,C_to_T,MTA1,homozygous_ref_ref,intron |
CONCEPT_CD as in Sample Data
Variant/Concept Name | Concept Code |
SNP | SO:0001483 |
indel | SO:1000032 |
INSTANCE_NUM
The set of all SNPs for each patient will all have the same encounter number and date. The concept codes will be the same for all SNPs (SO:0001483) and for all indels (SO:0001483). The set of all SNP facts will be enumerated in the instance_num field to make the primary key unique, as will the set of all indels.
VALTYPE_CD
This field will always equal “B” to indicate that data are stored in the observation_blob field and to trigger the full text search already existing in the i2b2 environment.
OBSERVATION_BLOB
<RSID | “missing_rsid”>,<REF_TO_ALT>,<GENE_SYMBOL | “missing_gene”>,<ZYGOSITY | “missing_zygosity”>,<CONSEQUENCE | “missing_consequence”>
{kind=link}